MA plot
DNA microarrays consist of an arrayed series of thousands of microscopic spots of DNA which allow comparisons between two samples of RNA or DNA (target samples), which can provide data on relative gene expression levels (in the case of RNA) or gene copy number (for DNA). The data obtained from two-color DNA microarrays come in the form of fluorescent Red (Cy5) and Green (Cy3) dye intensities. One-color oligonucleotide arrays use only a single fluorescent dye.
Microarray data is often normalized within arrays to control for systematic biases in dye coupling and hybridization efficiencies, as well as other technical biases in the DNA probes and the print tip used to spot the array[1]. By minimizing these systematic variations, true biological differences can be found. To determine whether normalization is needed, one can plot Cy5 (G) intensities against Cy3 (R) intensities and see whether the slope of the line is around 1. An improved method, which is basically a scaled, 45 degree rotation of the R vs. G plot is an MA-plot[2]. The MA-plot is a plot of the distribution of the red/green intensity ratio ('M') plotted by the average intensity ('A'). M and A are defined by the following equations.
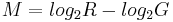
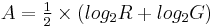
M is, therefore, the intensity ratio and A is the average intensity for a dot in the plot. MA plots are then used to visualize intensity-dependent ratio of raw microarray data. The MA plot uses M as the y-axis and A as the x-axis. The MA plot gives a quick overview of the distribution of the data. In many microarray gene expression experiments, the general assumption is that most of the genes would not see any change in their expression. Therefore the majority of the points on the y axis (M) would be located at 0, since Log(1) is 0. If this is not the case, then a normalization method such as LOESS should be applied to the data before statistical analysis.
The MA plot is the application of a Bland–Altman plot for visual representation of two channel DNA microarray gene expression data which has been transformed onto the M and A scale.
Contents |
Packages
Several Bioconductor packages, for the R software, provide the facility for creating MA plots, these include affy (ma.plot, mva.pairs), limma (plotMA), marray (maPlot), and edgeR(maPlot)
Similar "RA" plots can be generated using the raPlot function in the caroline CRAN R package.
Example in the R Programming Language
GeSHi Error: GeSHi could not find the language rsplus (using path /usr/share/php-geshi/geshi/) (code 2)
You need to specify a language like this: <source lang="html4strict">...</source>
Supported languages for syntax highlighting:
abap, actionscript, actionscript3, ada, apache, applescript, apt_sources, asm, asp, autoit, avisynth, bash, basic4gl, bf, bibtex, blitzbasic, bnf, boo, c, c_mac, caddcl, cadlisp, cfdg, cfm, cil, cmake, cobol, cpp, cpp-qt, csharp, css, d, dcs, delphi, diff, div, dos, dot, eiffel, email, erlang, fo, fortran, freebasic, genero, gettext, glsl, gml, gnuplot, groovy, haskell, hq9plus, html4strict, idl, ini, inno, intercal, io, java, java5, javascript, kixtart, klonec, klonecpp, latex, lisp, locobasic, lolcode, lotusformulas, lotusscript, lscript, lsl2, lua, m68k, make, matlab, mirc, modula3, mpasm, mxml, mysql, nsis, oberon2, objc, ocaml, ocaml-brief, oobas, oracle11, oracle8, pascal, per, perl, php, php-brief, pic16, pixelbender, plsql, povray, powershell, progress, prolog, properties, providex, python, qbasic, rails, rebol, reg, robots, ruby, sas, scala, scheme, scilab, sdlbasic, smalltalk, smarty, sql, tcl, teraterm, text, thinbasic, tsql, typoscript, vb, vbnet, verilog, vhdl, vim, visualfoxpro, visualprolog, whitespace, whois, winbatch, xml, xorg_conf, xpp, z80
See also
References
- ^ YH Yang, S Dudoit, P Luu, DM Lin, V Peng, J Ngai, TP Speed. (2002). Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Research vol. 30 (4) pp. e15.
- ^ Dudoit, S, Yang, YH, Callow, MJ, Speed, TP. (2002). Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Stat. Sin. 12:1 111-139